先天性サイトメガロウイルス感染児の「遅発性難聴」に対する抗ウイルス薬の医師主導治験が始まりました
治験の内容について
バルガンシクロビル塩酸塩ドライシロップの有効性および安全性を評価する
多施設共同プラセボ対照ランダム化並行群間比較試験(VGCV-3)
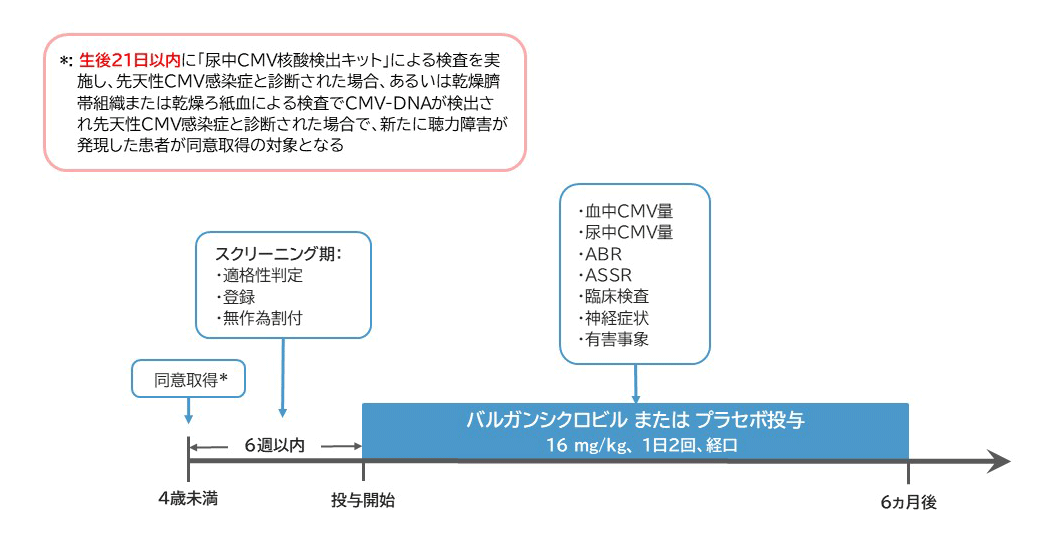
- 多施設共同、二重遮蔽、無作為割付、プラセボ対照、探索的試験です。
-
バルガンシクロビルの投与方法
バルガンシクロビル1回16 mg/kgを1日2回、経口投与します。 -
プラセボの投与方法
体重換算した被験薬(16 mg/kg)と同じ用量(mL)を1日2回、経口投与します。 - 投与期間:6ヵ月間
-
登録予定期間:2025年3月1日~2027年9月30日
バルガンシクロビル群10例、プラセボ群10例の計20例を目標としています。
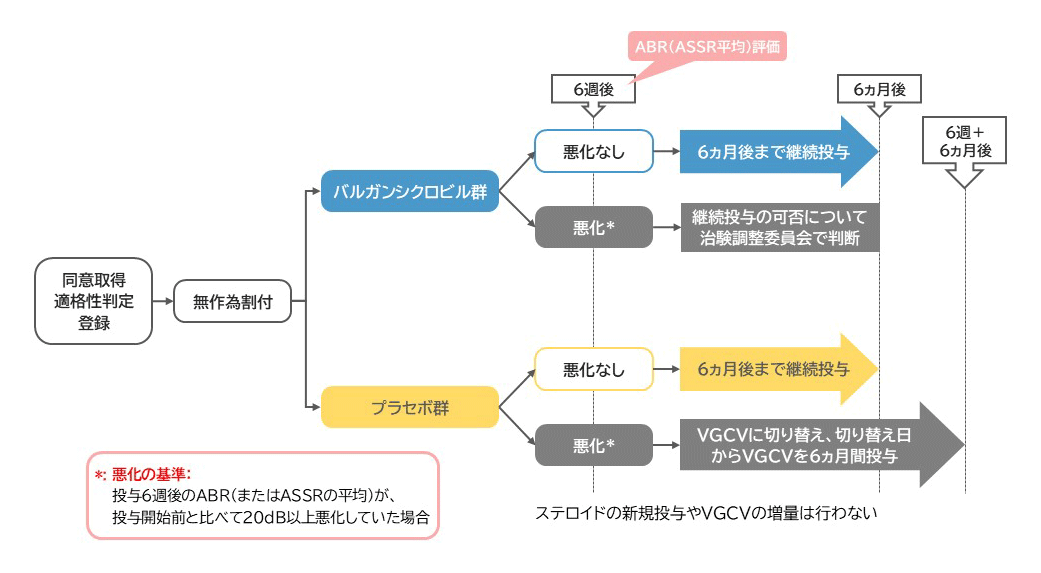
図:治験デザイン
先天性CMV感染症で遅発性難聴を呈した
患者さんが対象となります
Ⅰ. 対象となる患者さんの基準
① 生後21日以内に実施した尿中CMV核酸検出キットで先天性CMV感染症と診断された患者、あるいは、乾燥臍帯組織または乾燥ろ紙血による検査でCMV-DNAが検出され先天性 CMV 感染症と診断された患者
② 出生時は無症候性で生後21日以後に遅発性難聴※1を呈した患者、または出生時に症候性であっても聴覚障害を認めておらず、その後に遅発性難聴※1を呈した患者
③ 遅発性難聴の障害の程度が、少なくとも片耳で30dB以上の患者(スクリーニング時)
④ 満7歳未満(ただし就学前)
⑤ 本治験への登録前に治験の内容について十分な説明を行い、治験への参加に代諾者から文書による同意が得られた患者
※1:遅発性難聴とは、以下のいずれかを満たすものをいう。
・新生児聴覚スクリーニング検査で「正常」と判断され、その後生後21日以後の聴性脳幹反応(ABR)で少なくとも片耳で30dB以上と判断された場合、または、聴性定常反応(ASSR)の周波数(1000Hz、2000Hz、4000Hz)の平均値で少なくとも片耳で30dB以上と判断された場合
・新生児聴覚スクリーニング検査で「要再検」、その後生後21日以後のABRまたはASSRで「正常」と判断されたが、その後ABRまたはASSR(1000Hz、2000Hz、4000Hzの平均値)で少なくとも片耳で30dB以上と判断された場合
Ⅱ. 除外基準
① 抗生物質の投与が必要な細菌感染症の患者
② 血清クレアチニンが1.5mg/dLを超える患者
③ 他の原因の水頭症の患者
④ 前庭水管拡大などの内耳奇形や中耳奇形、真珠腫などの中耳疾患が認められる患者※2
⑤ ムンプス難聴などのCMV以外のウイルス性難聴が疑われる患者※3
⑥ その他、CMV以外の原因による難聴が明らかな患者※4
⑦ 過去にCMVに対する抗ウイルス薬(ガンシクロビルまたはバルガンシクロビル等)の治療を受けた患者。ただし、活動期病変や急性期病変に対する一時的な治療の場合は許容する。
⑧ 好中球数500/mm3未満または血小板数25,000/mm3未満の患者
⑨ 患児の母親がHIV陽性または患児がHIV陽性の患者※5
⑩ 抗ウイルス薬または免疫抑制薬を投与中の母親から母乳を投与されている患者
⑪ 同意取得前6ヵ月以内に他の治験薬の投与を受けたことがある、または治験参加期間に他の臨床試験に参加する予定がある患者
⑫ 治験責任医師または治験分担医師が本治験の対象として不適格であると判断した患者
※2:進行性難聴を呈する前庭水管拡大などの内耳奇形や中耳奇形、中耳真珠腫はCT画像検査などにより鑑別を行うことで区別する。また、滲出性中耳炎については、CTまたは耳鼻咽喉科での診察によって診断して区別する。
※3:ムンプス難聴については、病歴やムンプスIgMの血清学的診断により区別する。
※4:遺伝性難聴や中枢性進行性難聴などは病歴や病態で判断する。
※5:患児の母親がHIV陰性の場合には患児も陰性として扱う。
本治験で調べる内容について
Ⅰ. 有効性評価
(1)主要評価項目として、ベースライン時点の聴性脳幹反応(ABR)またはASSR(聴性定常反応)※6に基づき軽度以上の聴力障害を有するTotal earにおける、治験薬投与開始6ヵ月後の「聴力改善」の有無を調べます。
表1 聴力障害レベルの分類※7

ABR: Auditory Brainstem Response, ASSR: Auditory Steady-State Response
表2 聴力障害のベースラインからの変化の分類

【聴力改善の基準】
ABRまたはASSRに基づく聴力障害レベルのベースラインからの変化を、①改善、②不変(聴力は正常のまま)、③不変(聴力障害は同程度)、④増悪の4分類とし、「①改善」かつ聴力が10dB以上改善したものを「聴力改善」とする。
(2) 副次評価項目として、以下の項目を調べます。
1)ベースライン時点のABRまたはASSR※6に基づき軽度以上の聴力障害を有するTotal earにおける、治験薬投与開始6週後の「聴力改善」の有無
2)治験薬投与開始6週後および6ヵ月後の聴力障害レベルについて、ABRまたはASSR※6に基づくBest response earにおける以下の項目
・「聴力改善」の有無
3)治験薬投与開始6週後および6ヵ月後の聴力障害レベルについて、ABRに基づくTotal earにおける以下の項目
・「聴力改善」の有無(ベースライン時点のABRに基づき軽度以上の聴力障害を有するTotal earにおいて評価)
・①または②に該当する変化の有無
・①または②または③に該当する変化の有無
・ABR出現レベル(dB)のベースラインからの変化量
4)治験薬投与開始6週後および6ヵ月後の聴力障害レベルについて、ABRに基づくBest response earにおける以下の項目
・「聴力改善」の有無(ベースライン時点のABRに基づき、少なくとも片耳が軽度以上の聴力障害を有する症例において評価)
・①または②に該当する変化の有無
・①または②または③に該当する変化の有無
・ABR 出現レベル(dB)のベースラインからの変化量
5) ASSRに基づく各周波数※8の聴力障害レベルについて、ABRの 3)および4)と同様の項目
6) 全血中CMV量(治験薬投与6ヵ月時点のベースラインからの変化、経時推移)
7) 血漿中CMV量(治験薬投与6ヵ月時点のベースラインからの変化、経時推移)
8) 尿中CMV量(治験薬投与6ヵ月時点のベースラインからの変化、経時推移)
9) ベースラインで認められた血小板数減少、肝機能障害の改善
※6:適格性判定においてABRが正常でASSRにて30dB以上と判断され試験登録された症例については、ASSRに基づき聴力改善の判定を行う。
※7:ABRの評価は、中央判定委員会で実施する(ASSRの評価は測定機器の自動判定とする)。
※8:周波数は、500Hz、1000Hz、2000Hz、4000Hzとする。
Ⅱ. 安全性評価
有害事象や副作用がないかを調べます。
バルガンシクロビルの有効性と安全性を証明するために、こうした評価を行います。治験開始から6ヵ月の投与期間中は、定期的に受診していただくようお願いします。
難聴が悪化した場合の対応について
(被験者保護:バルガンシクロビルへの切り替え)
治験薬の投与6週後のABR(またはASSRの平均)が、投与開始前と比べて20dB以上悪化していた場合は、割付群の盲検解除を行い、以下の対応を行います。
Ⅰ. 被験薬群(バルガンシクロビル群)の場合
被験薬(バルガンシクロビル)の継続投与の可否について、治験調整委員会で被験者保護の観点から検討・決定します。
Ⅱ. 対照薬群(プラセボ群)の場合
被験者保護のため、対照薬(プラセボ)を被験薬(バルガンシクロビル)へ切り替えて投与開始します。切り替えは、原則として投与6週後の観察日から1ヵ月(30日)以内に実施します。被験薬の投与は、切り替え日(起算日)から6ヵ月間投与し、規定の観察・検査を行います。
図:被験者保護(バルガンシクロビル[VGCV]への切替)の手順